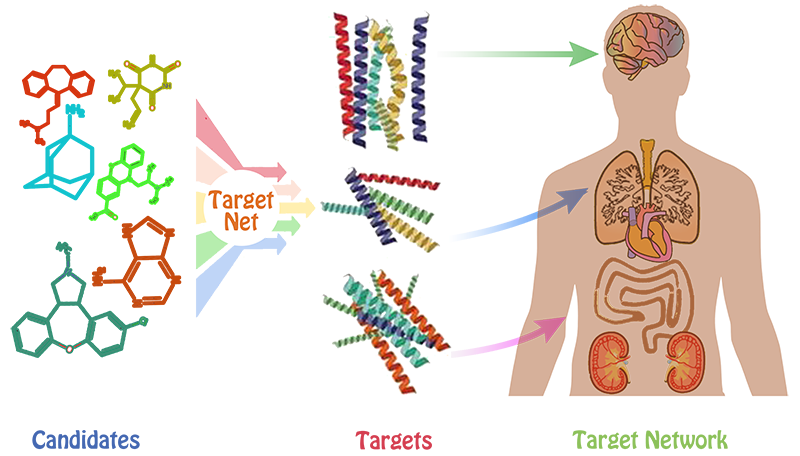
Drug-target interactions (DTIs) are central to current drug discovery processes and public health fields. In drug discovery process, one of the challenges is to identify the potential targets for drug-like compounds. Once the target is successfully identified, several receptor-based drug design methods could be easily used to optimize the structure of compounds, aims at improving the biological activity of these compounds.
However, this is usually a very difficult task for most of medicinal chemists. Furthermore, analyzing the DTI profiling of the drugs helps to infer drug indications, adverse drug reactions, drug-drug interactions, and drug mode of actions. Therefore, it is of high importance to reliably and fast predict DTI profiling of the drugs on a genome-scale level. Herein, we attempted to address the difficulty by developing a web server called TargetNet.
 TargetNet
is an open web server that could be used for
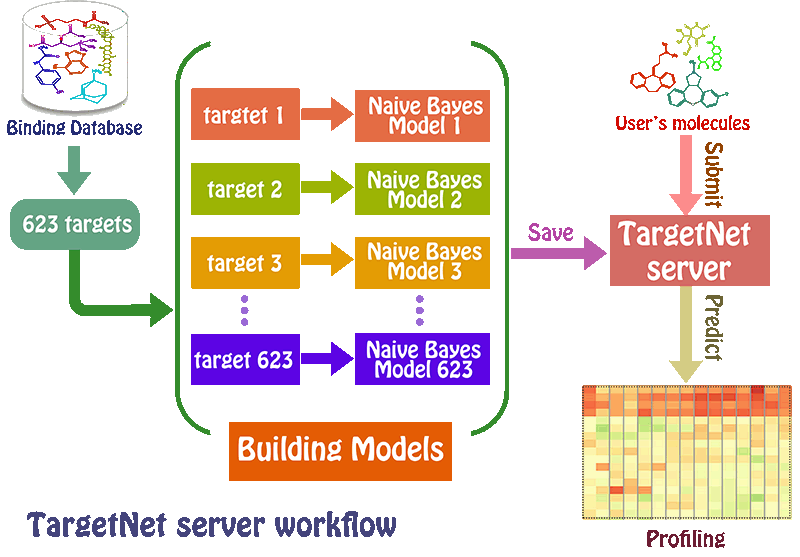
netting or predicting the binding of multiple targets for any given molecule. TargetNet simultaneously constructs a large number of QSAR models
based on current chemogenomics data to make future predictions. When the user submits a molecule, the server will predict the activity of the user’s
molecule across 623 human proteins by establishing the high quality QSAR model for each human protein, thus generating a DTI profiling that can used
as a feature vector for wide applications.
TargetNet
is an open web server that could be used for
netting or predicting the binding of multiple targets for any given molecule. TargetNet simultaneously constructs a large number of QSAR models
based on current chemogenomics data to make future predictions. When the user submits a molecule, the server will predict the activity of the user’s
molecule across 623 human proteins by establishing the high quality QSAR model for each human protein, thus generating a DTI profiling that can used
as a feature vector for wide applications.
The 623 QSAR models related to 623 human proteins were strictly evaluated and validated by several model
validation strategies, resulting in the AUC scores of 75%-100%. We applied the generated DTI profiling to successfully predict potential targets,
toxicity classification, drug-drug interactions, and drug mode of action, which sufficiently demonstrated the wide application value of the potential
DTI profiling.
- Zhi-Jiang Yao , Jie Dong , Yu-Jing Che , Min-Feng Zhu, Ming Wen, Ning-Ning Wang, Shan Wang, Ai-Ping Lu, Dong-Sheng Cao. TargetNet: a web service for predicting potential drug-target interaction profiling via multi-target SAR models. Journal of Computer-Aided Molecular Design, 2016 May 11. [PDF]